Identification of Novel Metabolic Hubs that Regulate Atherosclerotic Risk Factors

Figure 1: Fluorescently-labeled LDL uptake in human hepatic cells.
Epidemiological studies have identified many environmental and genetic factors that contribute to the development of atherosclerotic cardiovascular disease, the leading cause of morbidity and mortality worldwide. In particular, hypercholesterolemia has been identified as a major causal and modifiable risk factor for atherogenesis. As a result, substantial therapeutic progress has resulted from the widespread use of statins and other lipid-lowering drugs aimed at reducing plasma low-density lipoprotein (LDL)-cholesterol. Despite this, statins are not sufficient to prevent the progression of atherosclerosis in many individuals and in certain cases statin therapy is poorly tolerated.
The circulating levels of LDL are determined in large part by the rate of uptake through the hepatic LDL receptor (LDLR) , as evidenced by mutations in the genes encoding LDLR itself, its ligand apolipoprotein B, or its negative regulator PCSK9, which lead to the massive accumulation of plasma LDL in patients with familial hypercholesterolemia (FH). Although many pathways and genes involved in LDL uptake have been characterized throughout the years, surprisingly, our molecular understanding of LDL-cholesterol regulation remains incomplete. It is therefore likely that additional genes and pathways contribute to cholesterol regulation in humans.
Using an integrated genomic strategy in human hepatic cell lines (Fig. 1; Nature Med 2105), we recently identified an unexpected role for the adenosylcobalamin pathway in regulating LDLR expression and activity (Nature Comm 2021). Mechanistically, we demonstrated that loss of MMAB, a mitochondrial enzyme which catalyzes the conversion of vitamin B12 to adenosylcobalamin, controls LDLR activity through the methylmalonic and propionic acid-mediated inhibition of cholesterol biosynthesis and subsequent upregulation of SREBP2-mediated gene expression (Fig. 2). These findings extend the current knowledge of how intermediary metabolism relates to facets of mitochondrial signaling/biology and highlight the therapeutic potential of altering intracellular metabolites to treat CVD.

Figure 2: Schematic showing the role of the adenosylcobalamin pathway in regulating hepatic LDLR expression and circulating LDL-C levels. Scheme from PMID:3470386, Figure 8b.
Intriguingly, recent genetic data demonstrate that SNPs in the MVK/MMAB locus are strongly associated with altered plasma levels of HDL-C, total cholesterol and CVD risk in humans. While our previous work provides insight into the mechanisms by which MMAB variants may lead to altered plasma LDL-C levels, the mechanisms linking the adenosylcobalamin pathway to CVD biology remain incompletely understood. In light of these advances, our lab aims to uncover the causal and tissue-specific mechanism(s) by which MMAB influences cardiometabolic health. In addition, we seek to uncover additional metabolic hubs which regulate LDLR expression and activity by evaluating candidates from our genome-wide RNAi screen with known roles in intermediary metabolism and/or mitochondrial signaling (Fig. 3).
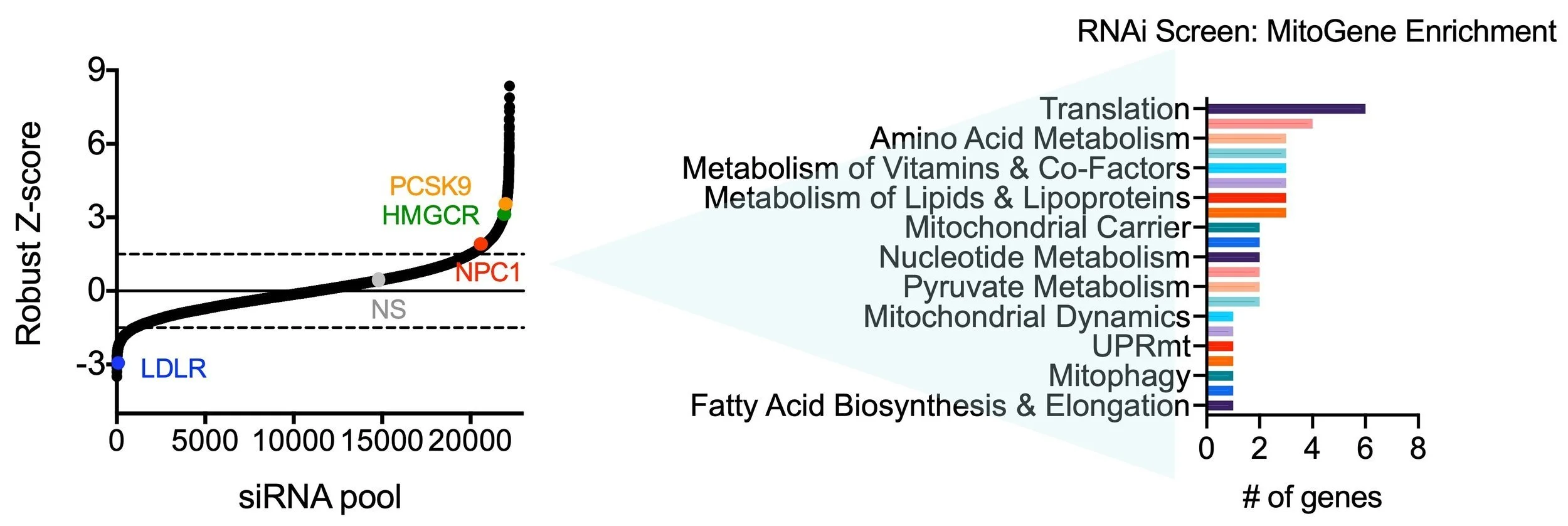
Figure 3: Gene enrichment analysis of top hits from the primary RNAi screen. Candidate genes whose loss-of-function increased or decreased LDLR activity were filtered for previously known roles in mitochondrial metabolism using MitoXplorer 1.0. Graphs modified from PMID:3470386, Figure 1.
Relevant Publications
Relevant Funding
Schneider-Lesser Foundation Fellowship for Junior Faculty | Regulation of Lipid Metabolism by UBOX5
ES-DRC Pilot & Feasibility Award | Cobalamin-Dependent Metabolism and Steatotic Liver Disease
AHA Transformational Project Award | Immunometabolic Regulation of Atherosclerosis by Methylmalonic Acid